大数据文摘作品,欢迎个人转发朋友圈,自媒体、媒体、机构转载务必申请授权,后台留言“机构名称+转载”,申请过授权的不必再次申请,只要按约定转载即可 选文|孙强 翻译整理|孙强 丁一 汪霞 作者|Barbara Han
2014年4月,就在世界卫生组织的官员们刚刚确定了一系列在几内亚可疑死亡案例是埃博拉病毒爆发,10名生态学家、4位兽医和一人类学家就前往几内亚一个名为Meliandou的小村庄。 他们是一个科学考察团,想搞清楚这个疾病爆发是如何开始的。一个叫埃米尔的2岁大的男孩是“零号病人”,他是如何感染上了埃博拉病毒?因为我们相信人们通过接触受感染动物而感染上了埃博拉病毒,生态学家一直在搜寻窝藏并传递病毒的动物宿主(通常它们自身并没有生病)。 随着每一种人畜共患的疾病如埃博拉病毒的新爆发,科学家竞相识别宿主,使公众卫生官员可以判定疾病的传染途径,也许能防止更多疾病从动物宿主流到的人类 “溢出事件”。这就是今天我们所说的处理爆发疫情的反应模型。 在Meliandou,埃博拉侦探采访了村民,研究了附近森林中的灵长类动物种群和收集网中捕捉的蝙蝠。在2014 年 12 月,他们发表了在当地孩子们经常一起玩的地方,有一群住在树洞里吃昆虫的蝙蝠;小埃米尔从蝙蝠身上感染了埃博拉病毒的假设。但是在队伍到达村庄之前,树已经着过火了,蝙蝠也没了,所以调查人员不能肯定地说。 上图:小村庄 下图:被烧毁的树 可能导致小埃米尔感染病毒的吃昆虫的蝙蝠
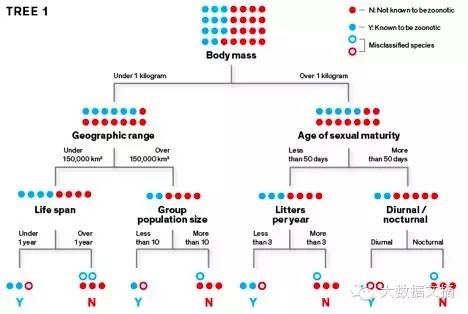
罪魁祸首:人类通过接触受感染的动物而感染人畜共患疾病。这种果蝠是尼帕病毒的已知载体,该病素导致致命的疾病,1999年首先在马来西亚确定。 过去大部分对埃博拉宿主的研究都集中在水果蝙蝠,该团队的发现则可能会促使科学家来研究这个食虫蝙蝠物种,并可能导致卫生官员在这些蝙蝠生活在接近人们居住的地区保持警惕。但是,这些都是应付一个残酷对手的防卫性动作:当前的埃博拉疫情在西非已造成超过11200人死亡,卫生官员仍在继续和它战斗。有没有办法变被动为主动,对抗埃博拉病毒和其他人畜共患病的进攻?我们能在疫情爆发前预测它,防患于未然吗? 在纽约州米尔布鲁克,卡里生态系统研究学院(CaryInstitute of Ecosystem Studies),我作为一名疾病生态学家用计算机建模和机器学习预测哪些野生物种能够引起未来的疾病爆发。我的模型创建可能宿主的“分类图”,揭示出了区分携带了对人体有害的微生物的不寻常的物种的功能参数组合。然后我用算法来整理数百或数以千计从未被检查的人畜共患疾病物种,基于其分类相似性计算任何给定的物种是疾病宿主的概率。这些模型给我们列出了一系列的嫌疑物种。 我和我的同事以科学探索精神做这个工作的,同时还有目前的紧迫感。传染性疾病在世界各地的兴起,据美国国际开发署的估计,新的疾病约75%是人畜共患病。如果我们能够预测哪些物种可能携带能够传播到人类的感染,我们就可以监控潜在的人们与这些动物互动的热点,。有一天,我希望生物学家能象气象学家预报天气那样预报疾病暴发。当然有一个显著的区别:一个气象学家不能阻止风暴发生,但我们也许能够阻止疫情。 机器是如何学习的 这张非常简单的示意图显示了我们怎样用算法创建分类树,然后利用该模型预测哪些啮齿动物携带人畜共患疾病。 学习树1:
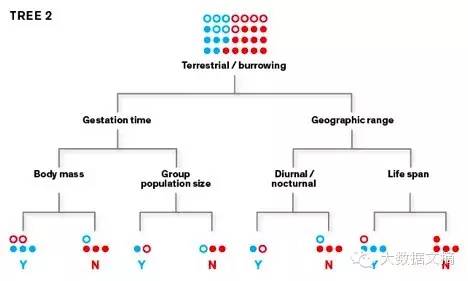
该算法使用训练数据集学习如何将物种分为“人畜共患”(这里用“Y”表示)或“不知道是否人畜共患”(这里用“N”来表示)。要创建初始分类树,它使用随机选择的参数(例如,低于或超过1公斤的体重大小),反复将啮齿动物的数据集分成两组。目标是在树的终端“叶” 层面上把 “Y” 从“N” 中分离出来。 学习树2:
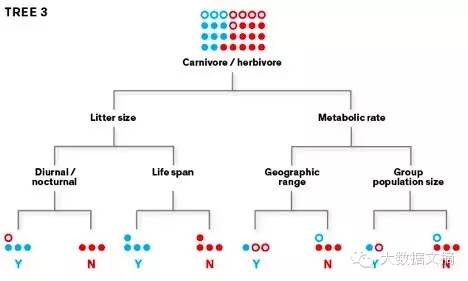
第一个学习树可能会产生大量分类错误,所以我们利用算法生成第二棵树,并将优先权赋予错误分类的物种,旨在正确地将它们重新分类。第二棵树错误分类的物种会在新建的第三个学习树中优先,以此类推。 学习树3:
以这种迭代的方式,该算法生成数千棵树。当数据通过所有这些树作为一个集合(ensemble)过滤,分类精度会逐渐提高。一旦模型在训练数据上表现良好,我们就用它来预测数据集的其余部分。 要理解为什么这种针对疾病爆发的被动反应方式迄今为止占上风,只要看一下埃博拉病毒。想象一下,如果你是一个野生动物学家,在刚果热带雨林(https://en.wikipedia.org/wiki/Congo_Basin)试图找到该病毒第一次出现的病原载体。你面前的森林和阿拉斯加差不多大小,有超过1400种哺乳动物和鸟类,以及数不清的昆虫种类。如果你有足够资源,你可以尝试取样每一种你能够捕捉的动物:很多是常见的物种,偶尔也有珍稀物种的零星标本。 即使是这样,你可能不会成功完成你的目标。一般只有一小部分病原物种的人口会被感染,并且可以看出,由于埃博拉暴发是间歇性的,病毒在它的动物宿主种群的感染被认为是非常低的。此外,可能有多个宿主物种 -而你需要在动态环境中把它们都找出来,包括动物的季节迁徙,以及由于栖息地遭到破坏的动物迁移。即使你拿到了感染的动物,你也不一定能从动物身上发现埃博拉病毒:病毒的数量在动物体内可随季节变化,或随着动物的身体遭受压力水平二变化。 在寻找埃博拉病毒的野生宿主的以往的调查中,生物学家们已经从几百个物种中收集了超过30,000个个体样本。虽然以前感染的痕迹(也就是抗体)已在若干动物的血液中检测到,我们尚未在活的动物的身体内分离出活病毒。生物学家不会放弃搜索,但显然是需要另外的方法时候了。 在我的工作中,我使用的机器学习算法,采用了大量野生动物的非结构化数据,并确定了那些是预测诉诸物种最有帮助的关键特征。我采用的算法工具是所谓的分类和回归树,人们已经使用了几十年。我的工作的创新之处在于,将这些技术应用于解决生态和全球健康的一个巨大的问题。 我最近在乔治亚大学和同事们进行的啮齿动物的研究,给出了这种方法如何工作的一个示例。啮齿目有超过2200多个种,多于其他任何哺乳动物。鼠类也携带多种病原体:据我们的保守估计,我们知道有大约200多种啮齿类动物能传播1到11种不等的人畜疾病。这其中包括可导致致命肺病的汉坦病毒,以及引起鼠疫的病菌,都给人类带来巨大苦难。 为了训练我们的算法来找到更多的病原菌载体,我们将80%数据输入模型,留下其余的数据作为检验数据集。我们给每个物种分配了二进制标签:“1” 表示该物种已知至少携带一种人畜共患疾病,或“0”,表示它的宿主状态未知。我们也收集了许多其它数据源,如有关哺乳动物的PanTHERIA数据库(http://esapubs.org/archive/ecol/E090/184/default.htm#abstract),它从数千有关啮齿类动物的实地研究中整理出各类数据, 如生理,行为,地域范围,社会结构等等。 该算法是这样创建分类树的 - 读取训练数据和识别分割点:能够将两个类别距离分地最远的参数的值。它一遍又一遍循环分割,创造分叉树枝,直到所有的数据被分类到一系列组,也就是分类树的叶子。它也可以创建一个回归树,这稍微有些技巧。其最后的叶子不只是展现二元分割点(如“每年产一窝”与“每年产超过一窝”);相反,它的叶子显示的是连续值(例如每年产1,2,3,4窝)。 在我们的研究中,该算法通过随机选择一个功能参数,将啮齿动物分成两个同质的亚组,由“1”和“0”来代表。虽然会尽可能做到准确,但难免有分类错误。然后,它选择的第二个特征参数,然后是第三个,以此类推,直到所有的啮齿动物已被分离到树的叶子。这些功能参数包括静止代谢率,身体尺寸,性成熟年龄,每窝子女数量,每年产几窝,群人口规模,以及其它共有超过50个功能特征。 去哪儿寻找病原菌载体
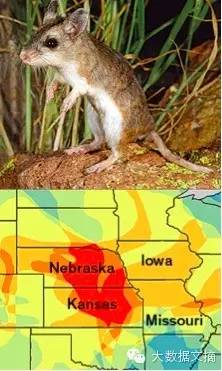
模型预测的疾病暴发热点 我们的计算机模型确定了58种以前从未记载过的人畜共患疾病的病原体,可能是感染人类的病菌的“藏身之地”。这些啮齿类动物的地理分布范围显示出两个潜在的热点,我们预测疾病暴发很可能发生在这些地方:美国中西部和横穿中亚和中东的一个带状地区。有了这些预测,实地生物学家可以实地考察研究,按照我们揭示的怀疑名单来一一研究,如北部蝗虫鼠(上图)和柽柳沙鼠(下图)。 北部蝗虫鼠(上图) 柽柳沙鼠(下图) 这种方法有一个很大的弱点:它对首先选择哪个功能参数非常敏感。取决于算法选择哪个参数,例如,选择“种群数量大小”或“代谢率”作为第一个功能参数,会产生非常不同的分类树。使用任何一棵树,我们都不会正确预测新的啮齿动物是否是人畜共患嫌疑;其预测准确率可能只比抛硬币好那么一点点。为了克服这种故障,我们采用了一个反复的过程称为“助推(boosting)”。通过“助推”,算法在下一轮建分类树重点考虑上一轮的错误,并优先考虑该数据。该方法会创造成百上千的分类树,虽然每棵树的预测能力很弱,当把它们做为一个集合来使用时,会产生一个非常准确的预测模型。 当我们用啮齿动物排序算法测试未被列入训练数据集的20%的啮齿动物时,预测物种是病菌宿主的准确率可以达到90%。而当我们拉去帷幕,回头审视哪些功能参数最利于准确预测,我们可以清楚看到,它是在一个特定的功能参数的集合的基础上来准确预测某动物的分类的。与你想象的可能不同,它并没有挑选出密切关联的物种。相反,它发现,这些病菌宿主有明显的“快速”的生命周期 -快速的增长率,性成熟早,频繁的抱窝产仔。这一发现能很好地吻合个别啮齿动物的深入研究,这些研究表明,宿主物种可能有较不敏感的免疫系统。这些动物可以忍受病原体,因为他们有一个“活得快,死得早”的策略: 他们的免疫系统不是他们的生活重点,因为他们只需要保持健康,活到能够繁殖下一代即可。分析结果还表明,宿主物种往往有较大的地理分部范围。这些动物可以生活在各种生态栖息地,也可以很好地适应人类创造的分散的、差别较大的环境。 我们的研究不仅仅是取得了一些科学见解:它还提供了可操作的智能信息。由于算法整理了2200啮齿类动物,它还产生了一个新的嫌疑清单。此前宿主状态未知的被分配为“0”的动物现在通过算法被分配到“1” -即已知是病原体宿主的一类。我们并不需要等待太久来验证我们的结果。在我们刚刚发布了我们的分析结果(http://spectrum.ieee.org/tech-通话/生物医学/诊断/计算机模型,identify-疾病轴承啮齿动物)后不久,我们清单上的两种嫌疑动物已经被认定为新的人畜疾病宿主。其中一个是红色的后盾田鼠(Myodes gapperi),原产于加拿大和美国北部,被发现携带导致虫病(echinococcosis)寄生虫,这是一种讨厌的疾病,病人会出现多器官囊肿。研究人员还确定了原产于小亚细亚的田鼠(Microtus guentheri)作为一个新发现的利什曼病(leishmaniasis)的宿主,该疾病导致皮肤溃烂。 我们的嫌疑名单给生物学家提供了新的机会:他们可以走到野外,进行“实地考察”来验证我们的研究结果。而这些实地的野外研究工作,反过来也会帮助我们的算法。随着实地研究的不断进展,生物学家会发现更多新的疾病携带者,我们的数据库将变得更富裕,我们的模型的预测精度将变得更加准确。该算法将继续发展,继续学习。 我们现在将我们的方法,以协助打击其他严重疾病。目前,我们正在努力,以确定哪些额外的蝙蝠可能是水库,导致出血热(https://en.wikipedia.org/wiki/Viral_hemorrhagic_fever)如埃博拉病毒和马尔堡病毒的丝状病毒。我们希望我们的研究结果将有助于解释蝙蝠如何一定可以忍受的感染是如此致命的大猿,包括人类。 我们的模型已经确定了一个蝙蝠种群系列,它们都在观察名单上。令我们惊讶的是,一些似乎能够携带埃博拉病毒的物种生活在非洲以外的国家,在这些地方从来没有人类暴发性出血热的正式报道。我们的结果给生物学家提出了一个问题:如果疫情真的没有在这些地方发生,为什么呢?这也是公共卫生官员应该考虑的问题:他们是否应该担心? 对于生态学,这门旨在了解数地球上数十亿复杂的、不断变化的生命体相互作用的学科,机器学习的方法具有有几个关键的优势。 例如,我们的算法可以处理我们的不完整的数据集。生物学家根本无法了解所有我迄今已经编目的1.6万个物种,更别说几百万我们都还没编目的。但是该算法考虑了任一物种的存在或缺失的数据,就像把它当做用于分类树的一个分割点的变量。 此外,我们的方法抵消了可能严重影响传染病研究的抽样偏差:在美国和欧洲这样富裕的地区的野生动物调查结果具有相对更高质量的数据。生物学家也可能在研究单个物种时失去对于偏见的警惕:他们更有可能会发现他们正在寻找的东西。比如说,他们发现挪威老鼠携带疾病X,他们很可能在该物种身上检查Y和Z疾病 ,结果导致某些物种很明显携带各种瘟疫,而很多物种还没有被检查是否有病原体。 我们的方法着眼于物种的固有特性,并最小化上述偏见的影响。例如,如果体型小的啮齿类动物在算法列为零,和它分为一组的小体型物种将遍布世界各地(因为体型小的啮齿动物有同样可能生活在贫国或富国)。通过使用物种内在生物学特征预测病原菌宿主状态,我们避免落入地域偏见的陷阱:只根据能负担调查监视的地方得到的数据开始预测。这样做,对于严重的数据缺乏,并没有太多的帮助。如果我们对一个物种没有数据,就不可能预测其(出现在)携带病菌的概率。我们的工作表明,削减基础研究经费有显著的连锁反应:不起眼的巴布亚新几内亚鼠的生活史的确是有价值去了解的。 机器学习也能很好地处理复杂性。生态分析经常包括几十个变量,但这些变量之间的相互作用往往不清楚。举例来说,虽然有证据显示动物的体型和代谢率之间的关系存在一个数学模型,但新生儿的大小和代谢率之间的关系就不那么清楚了。变数越多,就越难了解他们的复杂和隐蔽的相互作用。 但是,我们的算法并不需要我们对于这些相互作用设置任何规则。相反,我们的方法可以让数据自己说话。如果变量的特定组合导致极大地提高预测准确度,该模型能够识别这些变量,并向研究者显示以进行进一步的验证研究。该算法并不关心变量是如何相互作用的;它的唯一目标是最大限度地提升预测性能。这就需要我们自己的介入了。我们的任务是分析对预测最重要的变量,弄清他们揭示了关于人畜共患病的宿主的什么样的生物特征。 在向预测甚至抢先控制人畜共患疾病爆发的宏伟目标前进的过程中,只了解哪些疾病源于与那个生物库接触是不够的。生物学家需要明白:为什么这个物种特别?我们的算法给出了关于这个“为什么”的线索:生物学的机制使某些动物成为传播致死的传染病的载体。 当然,人类也在疾病的爆发中发挥作用,或者是直接与野生动物接触,或通过家畜与它们接触。例如,尼帕病毒在马来西亚出现源于人与感染的猪接触,猪与果蝠接触感染病毒。这些蝙蝠已经开始在果园和养猪场觅食,因为人们砍掉了他们的森林栖息地。 城市化,砍伐森林,和狩猎将继续使人类与潜在载有病毒的野生物种接触。我们同为这个生物圈的一部分,而疾病从这种复杂的系统中出现。我们才刚刚开始了解这些生态学动态。预测病原菌宿主是相当大的挑战,但我认为这只是更大挑战的一部分:搞清楚我们如何与野生动物和平共处,分享这个星球。 这篇文章首发于印刷版的《建立更好的疾病检测方法》。 原文链接:http://spectrum.ieee.org/biomedical/diagnostics/the-algorithm-thats-hunting-ebola 封面来源:huanqiukexue.com 关于作者 Barbara Han是一名Cary生态系统研究院(位于纽约州Millbrook)的科学家,虽然她在过去也做过生物实地考察,最近她通过她的数据集研究动物。她对于自己最终变成一个计算机程序员表示惊讶,但她对实地考察也并不充满怀念。“我喜欢与动物打交道,但我不喜欢被动物咬伤。 ”Han说。 大数据文摘医疗大数据栏目 2015年2月7日“医疗大数据专栏”正式成立。随着基因芯片及DNA测序技术的发展,生物医疗大数据迅猛发展,既是大数据技术发展的原动力,也是大数据技术的受益者。大数据文摘2013年7月成立,我们专注数据,每日坚持分享优质内容,从未间断,我们努力为读者和志愿者打造一个分享和交流平台。作为大数据文摘的志愿者群,我们中有不少人从事医疗大数据相关工作,关心医疗大数据的发展,愿意通过这个专栏,和大家一起分享医疗大数据的点点滴滴。 医疗大数据专栏主编介绍孙强,资深生物信息专家,现在服务于美国国立癌症研究所,从事癌症基因组数据库管理工作。热爱大数据,加入大数据文摘志愿者行列一年有余,愿以文会友,广结大数据善缘。旅居美国多年,现在定居于大华府地区。读过的学校:山东大学,中科院植物所,加大洛杉矶分校( UCLA )生活过的城市:淄博,济南,北京,洛杉矶,华盛顿其他爱好:足球,钓鱼,打牌 大数据格言:Big data is better data 译者简介 有意联系译者的朋友,请给“大数据文摘”后台留言,附自我介绍及微信ID,谢谢。 孙 强 资深生物信息专家,现在服务于美国国立癌症研究所,从事癌症基因组数据库管理工作。热爱大数据,加入大数据文摘志愿者行列一年有余,愿以文会友,广结大数据善缘。旅居美国多年,现在定居于大华府地区。 丁 一 杜克药理系博士在读,对生物信息学和相关的大数据挖掘很感兴趣。 汪 霞 A professional public health researcher with expertise in research synthesis andmodeling. I have successfully completed several projects with World Bank,European Union EU FP6, NIHR, and the British Society for Antimicrobial, WHO andUNICEF etc. in UK, Germany and China. |